ChemiQ是在量子计算机或虚拟机上模拟化学分子结构和性质的仿真软件——也是全球首款运用量子算法模拟的仿真软件——接入量子计算机计算速度呈指数增长。ChemiQ利用Jordan-Wigner,Parity等方法将二次量子化的Fermion的Hamiltonian算符转化(mapping)成Qubit的Hamiltonian算符(量子计算机识别的算符),算符间的转换是量子计算模拟化学过程的第一步,不同的转化方法对应着不同的Qubit信息,研究出所转化的算符少的mapping,相应的计算少,可大大简化计算;使用Unitary Coupled Cluster(简称UCC)等拟设构造模拟量子电路,分别代表不同的电路模型,所包含的参数数目和线路深度也不尽相同,构造出参数少、线路浅的线路拟设是量子计算模拟的关键,使得复杂的化学过程得到有效模拟;再结合量子相位评估(QPE),变分量子本征求解(VQE)算法,或量子虚时演化(QITE)算法模拟分子哈密顿量的期望值,进一步预测分子性质。这些算法不仅能保证量子态的相干性,其计算结果还能达到化学精度,在可预见的未来,有着极大的应用前景和优势。

图附3.1.1 ChemiQ软件
3.1 ChemiQ化学软件的安装¶
1、登录本源量子云官网图附3.1.1.png(https://qcloud.originqc.com.cn/),选择“应用推广云”,如图附3.1.2 所示;选择“生物医药”方向,点击“了解详情”如图附3.1.3所示;点击“Windows进行下载”,如图附3.1.4所示。(直达页面:https://qcloud.originqc.com.cn/chemistryIntroduce)

图附3.1.2 选择“应用推广云”

图附3.1.3 选择“生物医药”
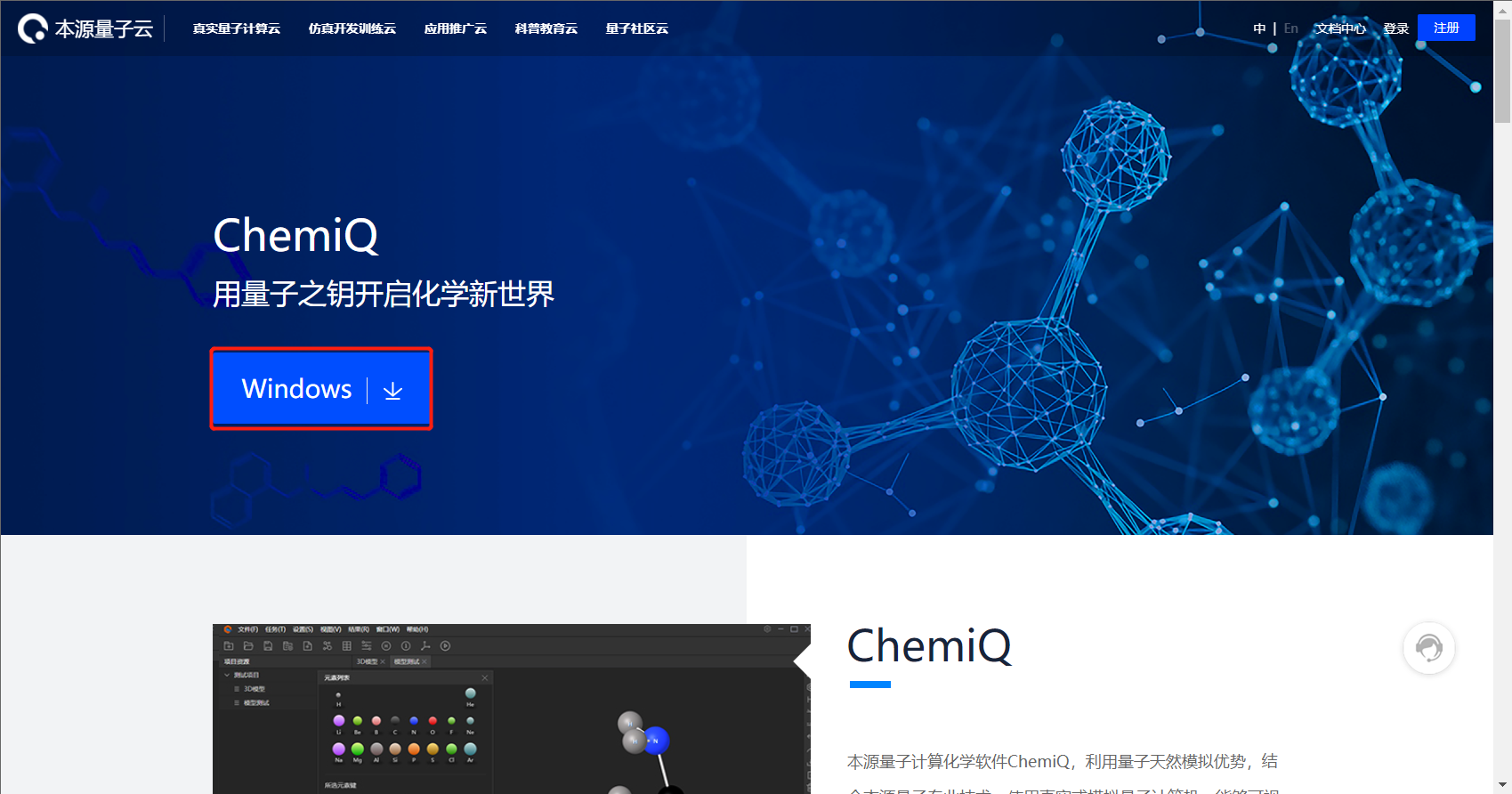
图附3.1.4 下载
2、下载完成后,双击安装该软件,点击“我同意”;
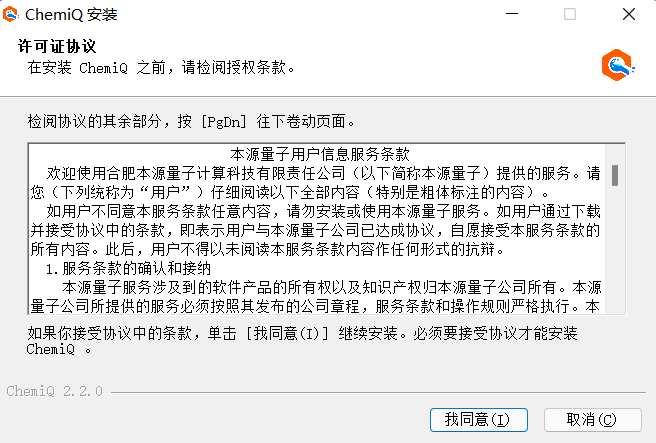
图附3.1.5 点击“我同意”
3、然后点击“下一步”;
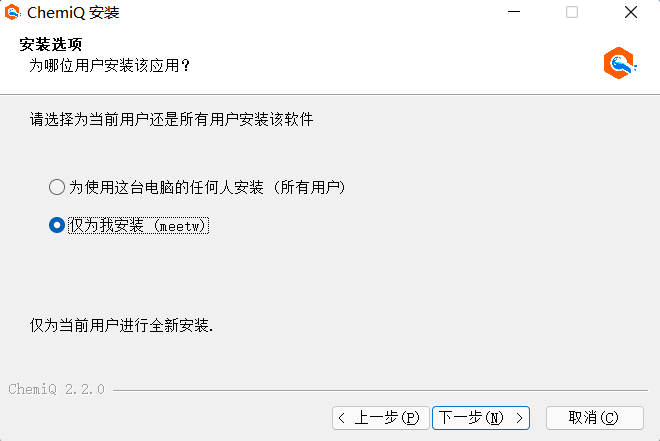
图附3.1.6 下一步
4、再安装到默认目录,点击“安装”;
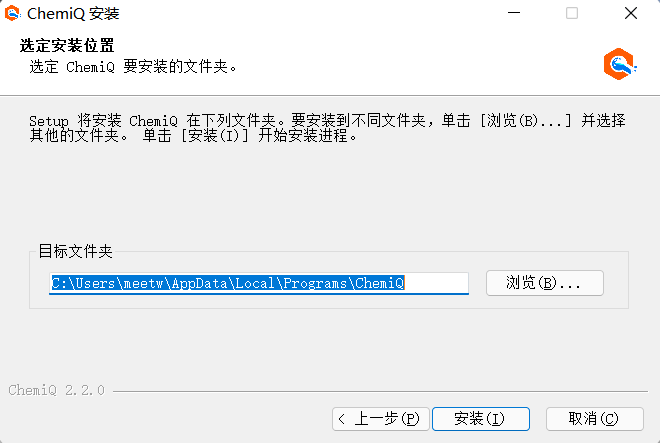
图附3.1.7 点击“安装”
5、安装完成后,运行该软件。

图附3.1.8 安装完成
3.2 ChemiQ软件应用示例¶
1、 新建项目。先新建一个项目名称——test H2,然后是填好创建人、计算模式、保存路径和项目描述,最后点击确定。
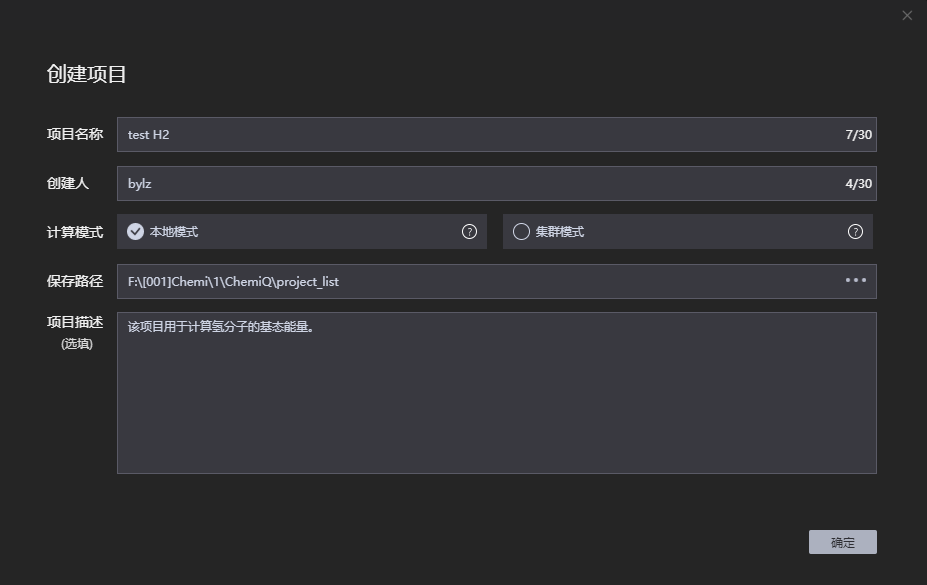
图附3.2.1 新建项目
2、 主界面。该软件左侧显示的是项目下的任务列表;左下显示的是项目或者任务详情;右侧显示分为上下两部分,上部分为分子模型,下部分为结果展示,可上下拖动调节大小。
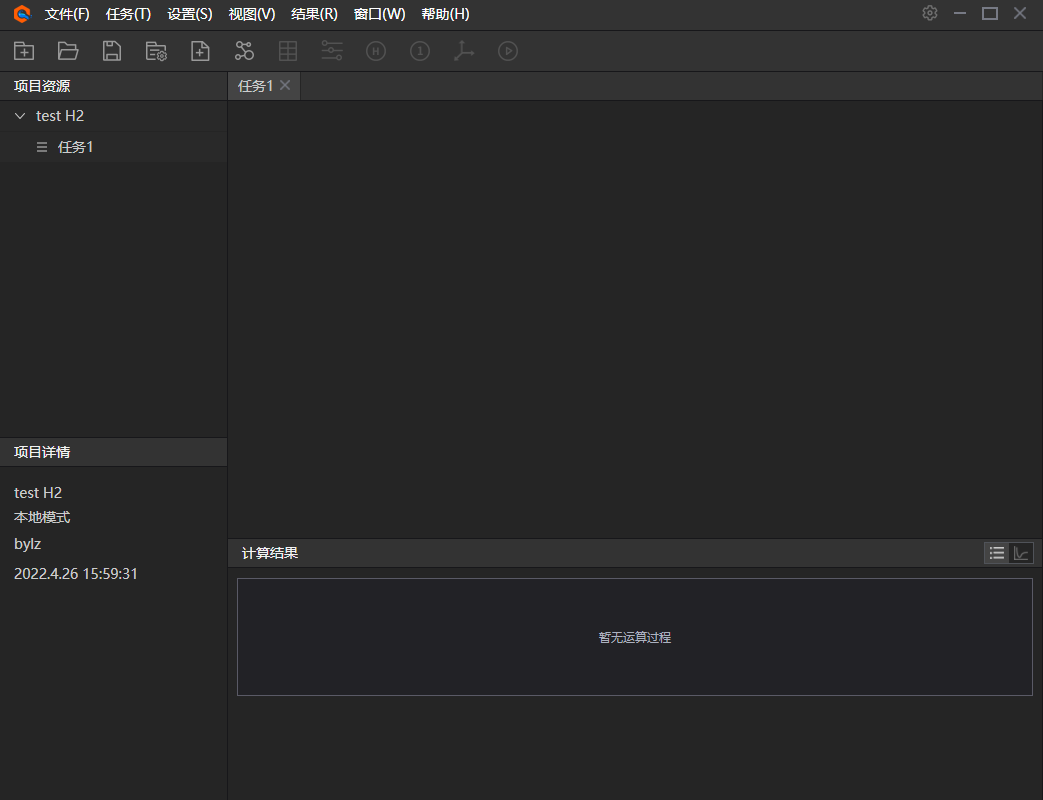
图附3.2.2 主页面
3、 分子建模。以氢分子势能曲线为例,首先是构建分子模型。点击设置-分子模型,或者工具栏中图标构建分子模型。在这里可使用构建分子快捷工具,如右侧弹出框。
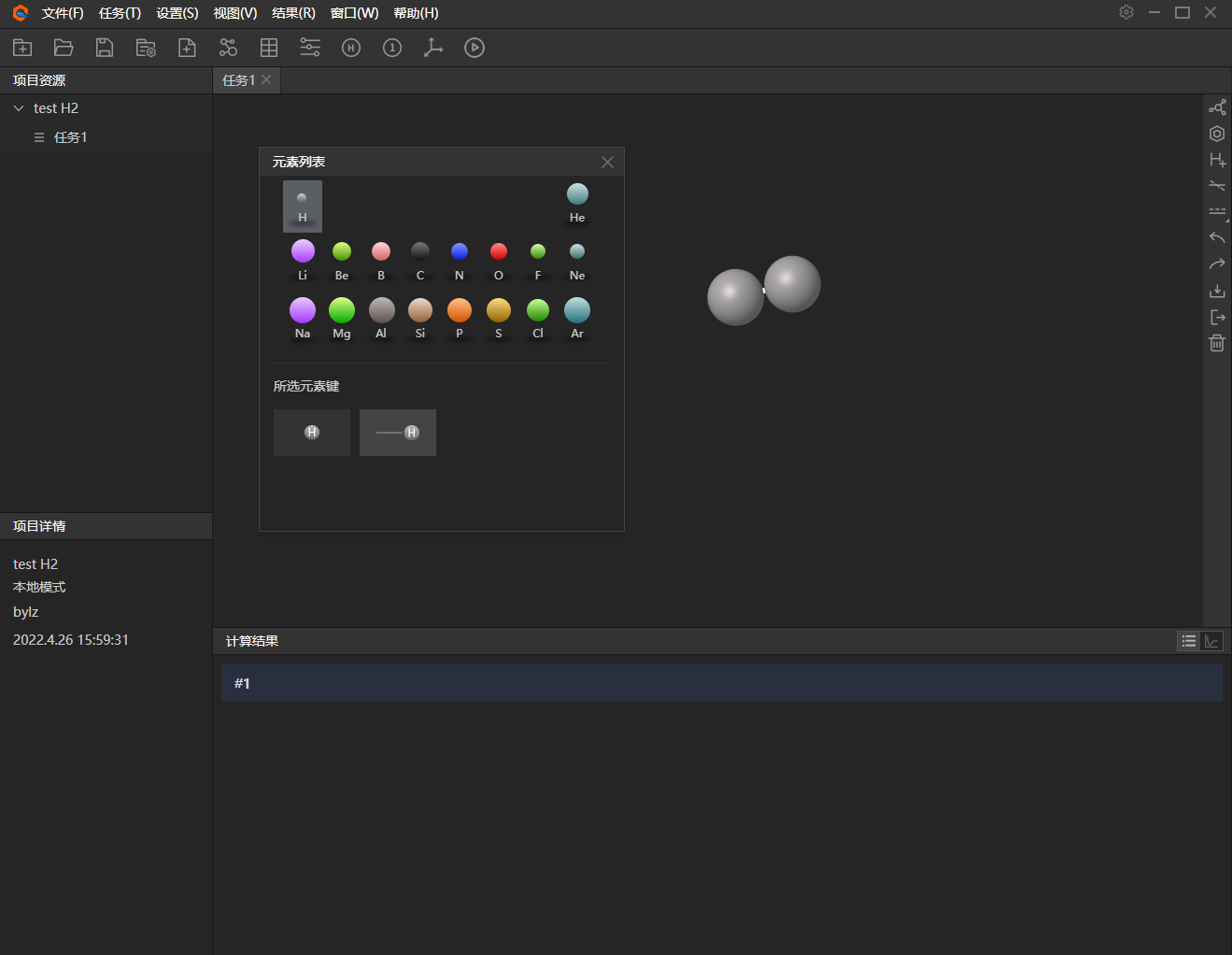
图附3.2.3 分子建模
4、 配置参数。在参数配置框中选择计算类型——势能曲线,基组、电荷、自旋多重度可根据用户对应设置,在这里以STO-3G基组模拟氢分子PES;扫描坐标选择氢分子中键长距离为变量,设置对应的节点个数和扫描区间;映射、拟设和优化器对应选择如下图所示:
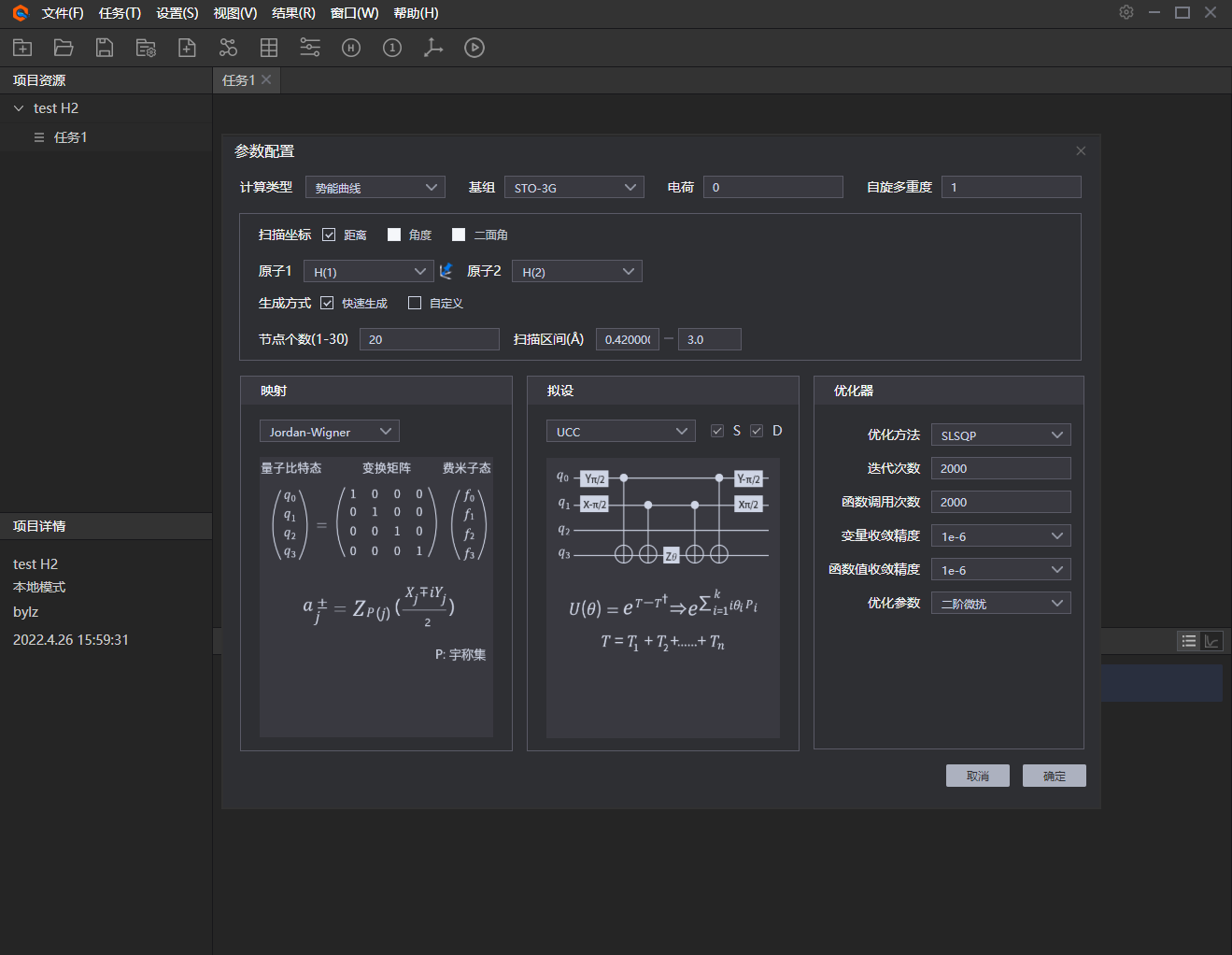
图附3.2.4 配置参数
5、任务详情。左下部分显示任务的参数详情,如计算结果中展示的计算列表,其中往下拉会显示20个节点任务;此外为了便于显示,可点击视图中便捷工具,如图展示是分子模型中的元素名称和元素编号。
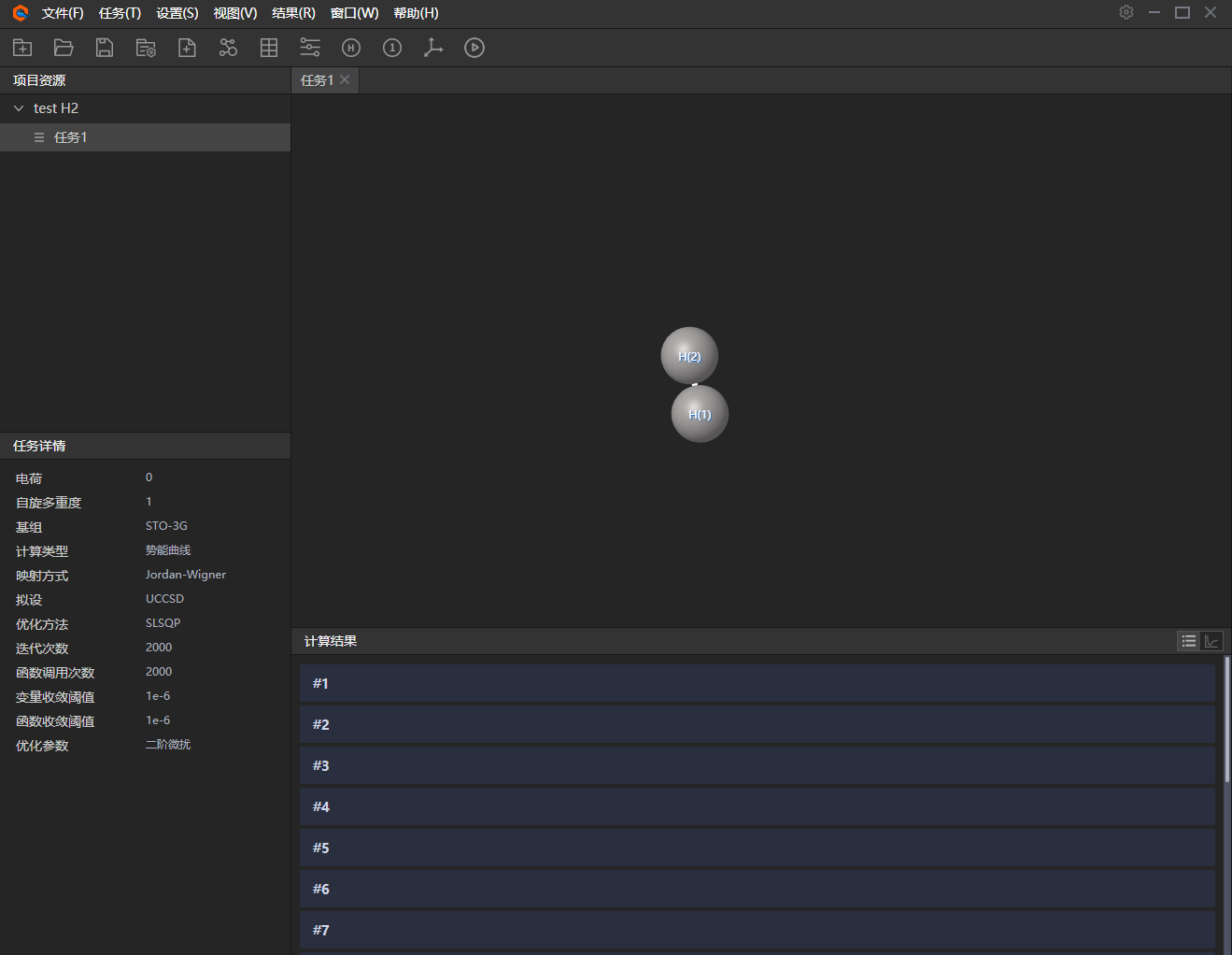
图附3.2.5 任务详情
6、 计算完成。可以看到计算结果已经展示出来,同时也可以看到每个氢分子坐标计算得到能量如下:

图附3.2.6 计算完成
7、 结果展示。势能曲线中可点击单个节点进入结果详情中,或者点击计算列表/势能曲线切换结果展示。
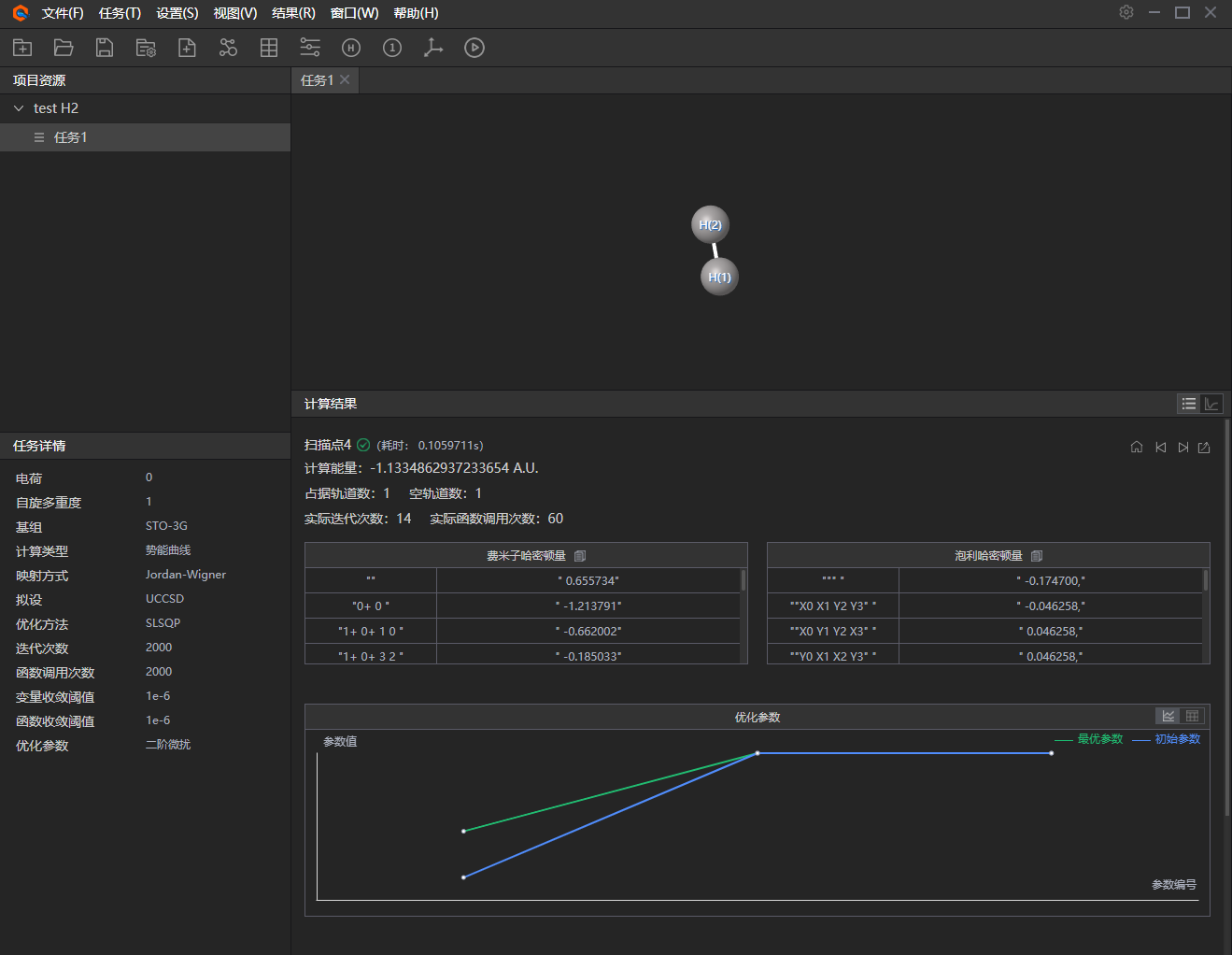
图附3.2.7 结果展示
3.3 ChemiQ接口介绍与使用¶
使用封装的ChemiQ计算接口进行实现
表附3.3.1,列出的是ChemiQ封装的计算接口:
表附3.3.1
接口名称 |
描述 |
initialize |
初始化量子化学计算的环境 |
finalize |
释放量子化学计算的环境 |
setMolecule |
设置单个分子模型 |
setMolecules |
设置一组分子模型 |
setMultiplicity |
设置重数 |
setCharge |
设置电荷数 |
setBasis |
设置计算基 |
setTransformType |
设置费米子到泡利算子的转换类型 |
setUccType |
设置UCC模型类型 |
setOptimizerType |
设置优化器类型 |
setOptimizerIterNum |
设置优化器迭代次数 |
setOptimizerFuncCallNum |
设置优化器函数调用次数 |
setOptimizerXatol |
设置优化器参数收敛阈值 |
setOptimizerFatol |
设置优化器函数收敛阈值 |
setLearningRate |
设置学习率 |
setEvolutionTime |
设置演化时间 |
setHamiltonianSimulationSlices |
设置哈密顿量模拟切片数 |
setSaveDataDir |
设置中间数据存放目录 |
setRandomPara |
设置随机优化参数 |
setDefaultOptimizedPara |
设置默认优化参数 |
setToGetHamiltonianFromFile |
设置从文件获取体系哈密顿量 |
setHamiltonianGenerationOnly |
设置只生成体系哈密顿量 |
exec |
执行计算 |
getLastError |
获取最后一条错误日志 |
initialize接口,作用是用来初始化量子化学计算环境,它需要传入一个变量就是量子化学计算包的路径,这里已经把PSi4安装在了python能检索到的环境路径下,使用时只需要传入空的字符串即可。 下面演示一下如何使用ChemiQ计算接口来实现氢分子的基态能量计算。
首先构造一组不同距离下的氢分子模型;然后生成ChemiQ的一个实例,调用setMolecules接口设置一组氢分子模型,设置氢分子的电荷数为0,自旋多重度为1;使用的计算基是sto-3g;UCC模型我们使用的是UCCS,费米子哈密顿量到泡利哈密顿量的转换这里用的是JW变换;这里使用的优化器是Nelder-Mead,优化器迭代次数为200,函数调用次数为200,显示优化器计算的中间结果;最后执行计算。
1.import matplotlib.pyplot as plt
2.
3.from pyqpanda import *
4.
5.if __name__=="__main__":
6.
7. distances = [x * 0.1 for x in range(2, 25)]
8. molecule = "H 0 0 0\nH 0 0 {0}"
9.
10. molecules = []
11. for d in distances:
12. molecules.append(molecule.format(d))
13.
14. chemiq = ChemiQ()
15. chemiq.initialize("")
16. chemiq.setMolecules(molecules)
17. chemiq.setCharge(0)
18. chemiq.setMultiplicity(1)
19. chemiq.setBasis("sto-3g")
20. chemiq.setUccType(UccType.UCCS)
21. chemiq.setTransformType(TransFormType.Jordan_Wigner)
22. chemiq.setOptimizerType(OptimizerType.NELDER_MEAD)
23. chemiq.setOptimizerIterNum(200)
24. chemiq.setOptimizerFatol(200)
25. chemiq.exec()
26. chemiq.finalize()
27.
28. value = chemiq.getEnergies()
29.
30. plt.plot(distances , value, 'r')
31. plt.xlabel('distance')
32. plt.ylabel('energy')
33. plt.title('VQE PLOT')
34. plt.show()
获取优化后的能量,绘制曲线图。这条曲线就是优化得到的氢分子在不同距离下对应的基态能量:
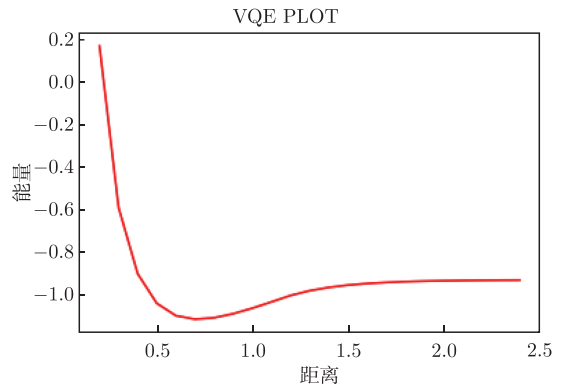
图附3.3.1 氢分子在不同距离下对应的基态能量
3.4 非梯度下降法实现VQE算法代码示例¶
首先,导入pyQPanda和psi4_wrapper中的所有模块,以及一些其它组件模块准备。
1.from pyqpanda import *
2.from psi4_wrapper import *
3.import numpy as np
4.from functools import partial
5.from math import pi
6.import matplotlib.pyplot as plt
然后,定义非梯度下降优化器使用的损失函数为loss_func,loss_func接受的一组参数为待优化的参数列表,轨道个数,电子个数,体系哈密顿量。这个接口是先用ccsd模型构造的费米子哈密顿量,然后利用JW变换将费米子哈密顿量转换为泡利哈密顿量,再接着将CC转化成UCC,再计算体系哈密顿量在试验态下的期望,最后返回期望值来实现的。
1.def loss_func(para_list, qubit_number, electron_number, Hamiltonian):
2. '''
3. <𝜓^∗|𝐻|𝜓>, Calculation system expectation of Hamiltonian in experimental state.
4. para_list: parameters to be optimized
5. qubit_number: qubit number
6. electron_number: electron number
7. Hamiltonian: System Hamiltonian
8. '''
9. fermion_cc =get_ccsd(qubit_number, electron_number, para_list)
10. pauli_cc = JordanWignerTransform(fermion_cc)
11. ucc = cc_to_ucc_hamiltonian(pauli_cc)
12. expectation=0
13. for component in Hamiltonian:
14. expectation+=get_expectation(qubit_number, electron_number, ucc, component)
15. expectation=float(expectation.real)
16. print(expectation)
17. return ("", expectation)
下面将对loss_func使用到的接口逐个进行讲解:
get_ccsd_n_term接口的作用是返回构造CCSD模型需要用到的参数个数,这个接口接收的参数是轨道个数和电子个数。
1.def get_ccsd_n_term(qn, en):
2. '''
3. coupled cluster single and double model.
4. e.g. 4 qubits, 2 electrons
5. then 0 and 1 are occupied,just consider 0->2,0->3,1->2,1->3,01->23
6. '''
7.
8. if n_electron>n_qubit:
9. assert False
10.
11. return int((qn - en) * en + (qn - en)* (qn -en - 1) * en * (en - 1) / 4)
get_ccsd接口则是用来构造普通参数对应的CCSD模型费米子哈密顿量,该接口接收的参数是轨道个数、电子个数和单激发双激发前面对应的系数。
1.def get_ccsd(qn, en, para):
2. '''
3. get Coupled cluster single and double model.
4. e.g. 4 qubits, 2 electrons
5. then 0 and 1 are occupied,just consider 0->2,0->3,1->2,1->3,01->23.
6. returned FermionOperator like this:
7. { {"2+ 0":var[0]},{"3+ 0":var[1]},{"2+ 1":var[2]},{"3+ 1":var[3]},
8. {"3+ 2+ 1 0":var[4]} }
9.
10. '''
11. if n_electron>n_qubit:
12. assert False
13. if n_electron==n_qubit:
14. return FermionOperator()
15.
16. if get_ccsd_n_term(qn, en) != len(para):
17. assert False
18.
19. cnt = 0
20. fermion_op = FermionOperator()
21. for i in range(en):
22. for ex in range(en, qn):
23. fermion_op += FermionOperator(str(ex) + "+ " + str(i), para[cnt])
24. cnt += 1
25.
26. for i in range(n_electron):
27. for j in range(i+1,n_electron):
28. for ex1 in range(n_electron,n_qubit):
29. for ex2 in range(ex1+1,n_qubit):
30. fermion_op += FermionOperator(
31. str(ex2)+"+ "+str(ex1)+"+ "+str(j)+" "+str(i),
32. para[cnt]
33. )
34. cnt += 1
35.
36. return fermion_op
JordanWignerTransform接口的作用是将费米子哈密顿量转换成泡利哈密顿量。
1.def JordanWignerTransform(fermion_op):
2. data = fermion_op.data()
3. pauli = PauliOperator()
4. for i in data:
5. pauli += get_fermion_jordan_wigner(i[0][0])*i[1]
6. return pauli
get_fermion_jordan_wigner接口则是将费米子哈密顿量的子项转换成泡利哈密顿量。
1.def get_fermion_jordan_wigner(fermion_item):
2. pauli = PauliOperator("", 1)
3.
4. for i in fermion_item:
5. op_qubit = i[0]
6. op_str = ""
7. for j in range(op_qubit):
8. op_str += "Z" + str(j) + " "
9.
10. op_str1 = op_str + "X" + str(op_qubit)
11. op_str2 = op_str + "Y" + str(op_qubit)
12.
13. pauli_map = {}
14. pauli_map[op_str1] = 0.5
15.
16. if i[1]:
17. pauli_map[op_str2] = -0.5j
18. else:
19. pauli_map[op_str2] = 0.5j
20.
21. pauli *= PauliOperator(pauli_map)
22.
23. return pauli
cc_to_ucc_hamiltonian接口的作用是CC模型对应的哈密顿量转成UCC模型对应的哈密顿量。
1.def cc_to_ucc_hamiltonian(cc_op):
2. '''
3. generate Hamiltonian form of unitary coupled cluster
4. based on coupled cluster,H=1j*(T-dagger(T)),
5. then exp(-iHt)=exp(T-dagger(T))
6. '''
7. return 1j*(cc_op-cc_op.dagger())
get_expectation接口,作用是计算体系哈密顿量在试验态下的期望,接收的参数是轨道个数,电子个数,UCC模型,体系哈密顿量的一个子项。
1.def get_expectation(n_qubit, n_en, ucc,component):
2. '''
3. get expectation of one hamiltonian.
4. n_qubit: qubit number
5. n_en: electron number
6. ucc: unitary coupled cluster operator
7. component: paolioperator and coefficient,e.g. ('X0 Y1 Z2',0.2)
8. '''
9.
10. machine=init_quantum_machine(QMachineType.CPU)
11. q = machine.qAlloc_many(n_qubit)
12. prog=QProg()
13.
14. prog.insert(prepareInitialState(q, n_en))
15. prog.insert(simulate_hamiltonian(q, ucc, 1.0, 4))
16.
17. for i, j in component[0].items():
18. if j=='X':
19. prog.insert(H(q[i]))
20. elif j=='Y':
21. prog.insert(RX(q[i],pi/2))
22.
23. machine.directly_run(prog)
24. result=machine.get_prob_dict(q, select_max=-1)
25. machine.qFree_all(q)
26.
27. expectation=0
28. #奇负偶正
29. for i in result:
30. if parity_check(i, component[0]):
31. expectation-=result[i]
32. else:
33. expectation+=result[i]
34. return expectation*component[1]
prepareInitialState接口的作用是制备初态,接收的参数是一组量子比特和电子个数。
1.def prepareInitialState(qlist, en):
2. '''
3. prepare initial state.
4. qlist: qubit list
5. en: electron number
6. return a QCircuit
7. '''
8. circuit = QCircuit()
9. if len(qlist) < en:
10. return circuit
11.
12. for i in range(en):
13. circuit.insert(X(qlist[i]))
14.
15. return circuit;
simulate_hamiltonian接口,作用是构造哈密顿量的模拟线路,接收的参数是一组量子比特,泡利哈密顿量、演化时间演化次数。
1.def simulate_hamiltonian(qubit_list,pauli,t,slices=3):
2. '''
3. Simulate a general case of hamiltonian by Trotter-Suzuki
4. approximation. U=exp(-iHt)=(exp(-i H1 t/n)*exp(-i H2 t/n))^n
5. '''
6. circuit =QCircuit()
7.
8. for i in range(slices):
9. for op in pauli.data():
10. term = op[0][0]
11. circuit.insert(
12. simulate_one_term(
13. qubit_list,
14. term, op[1].real,
15. t/slices
16. )
17. )
18.
19. return circuit
simulate_one_term是构造哈密顿量子项的模拟线路。
1.def simulate_one_term(qubit_list, hamiltonian_term, coef, t):
2. '''
3. Simulate a single term of Hamilonian like "X0 Y1 Z2" with
4. coefficient and time. U=exp(-it*coef*H)
5. '''
6. circuit =QCircuit()
7.
8. if not hamiltonian_term:
9. return circuit
10.
11. transform=QCircuit()
12. tmp_qlist = []
13. for q, term in hamiltonian_term.items():
14. if term is 'X':
15. transform.insert(H(qubit_list[q]))
16. elif term is 'Y':
17. transform.insert(RX(qubit_list[q],pi/2))
18.
19. tmp_qlist.append(qubit_list[q])
20.
21. circuit.insert(transform)
22.
23. size = len(tmp_qlist)
24. if size == 1:
25. circuit.insert(RZ(tmp_qlist[0], 2*coef*t))
26. elif size > 1:
27. for i in range(size - 1):
28. circuit.insert(CNOT(tmp_qlist[i], tmp_qlist[size - 1]))
29. circuit.insert(RZ(tmp_qlist[size-1], 2*coef*t))
30. for i in range(size - 1):
31. circuit.insert(CNOT(tmp_qlist[i], tmp_qlist[size - 1]))
32.
33. circuit.insert(transform.dagger())
34.
35. return circuit
paity_check是对量子态中指定比特1的个数做奇偶校验。
1.def parity_check(number, terms):
2. '''
3. pairty check
4. number: quantum state
5. terms: a single term of PauliOperator, like"[(0, X), (1, Y)]"
6. '''
7. check=0
8. number=number[::-1]
9. for i in terms:
10. if number[i]=='1':
11. check+=1
12. return check%2
optimize_by_no_gradient是非梯度下降优化算法的主体接口,需要传入的一组参数是体系哈密顿量,轨道个数,电子个数,优化器迭代次数
接口的具体实现步骤是:首先初始化一组待优化的参数,然后构造一个非梯度下降优化器,这里构造的优化器是Nelder-Mead,设置优化器的迭代次数并向优化器注册计算期望的损失函数,然后执行优化器,最后返回优化器优化的最低期望值。
1.def optimize_by_no_gradient(mol_pauli, n_qubit, n_en, iters):
2. n_para = get_ccsd_n_term(n_qubit, n_electron)
3.
4. para_vec = []
5. for i in range(n_para):
6. para_vec.append(0.5)
7.
8. no_gd_optimizer = OptimizerFactory.makeOptimizer(OptimizerType.NELDER_MEAD)
9. no_gd_optimizer.setMaxIter(iters)
10. no_gd_optimizer.setMaxFCalls(iters)
11. no_gd_optimizer.registerFunc(partial(
12. loss_func,
13. qubit_number = n_qubit,
14. electron_number = n_en,
15. Hamiltonian=mol_pauli.toHamiltonian(1)),
16. para_vec)
17.
18. no_gd_optimizer.exec()
19. result = no_gd_optimizer.getResult()
20. print(result.fun_val)
21.
22. return result.fun_val
etAtomElectronNum接口作用是返回原子对应的电子个数。
1.def getAtomElectronNum(atom):
2. atom_electron_map = {
3. 'H':1, 'He':2, 'Li':3, 'Be':4, 'B':5, 'C':6, 'N':7, 'O':8, 'F':9, 'Ne':10,
4. 'Na':11, 'Mg':12, 'Al':13, 'Si':14, 'P':15, 'S':16, 'Cl':17, 'Ar':18
5. }
6.
7. if (not atom_electron_map.__contains__(atom)):
8. return 0
9.
10. return atom_electron_map[atom]
该算法演示示例对应的主函数,首先构造一组不同距离下的氢分子模型,然后计算每个氢分子模型对应的基态能量,最后将计算的结果绘制成曲线图。
1.if __name__=="__main__":
2. distances = [x * 0.1 for x in range(2, 25)]
3. molecule = "H 0 0 0\nH 0 0 {0}"
4.
5. molecules = []
6. for d in distances:
7. molecules.append(molecule.format(d))
8.
9. chemistry_dict = {
10. "mol":"",
11. "multiplicity":1,
12. "charge":0,
13. "basis":"sto-3g",
14. }
15.
16. energies = []
17.
18. for d in distances:
19. mol = molecule.format(d)
20.
21. chemistry_dict["mol"] = molecule.format(d)
22. data = run_psi4(chemistry_dict)
23. #get molecule electron number
24. n_electron = 0
25. mol_splits = mol.split()
26. cnt = 0
27. while (cnt < len(mol_splits)):
28. n_electron += getAtomElectronNum(mol_splits[cnt])
29. cnt += 4
30.
31. fermion_op = parsePsi4DataToFermion(data[1])
32. pauli_op = JordanWignerTransform(fermion_op)
33.
34. n_qubit = pauli_op.getMaxIndex()
35.
36. energies.append(optimize_by_no_gradient(pauli_op, n_qubit, n_electron, 200))
37.
38. plt.plot(distances , energies, 'r')
39. plt.xlabel('distance')
40. plt.ylabel('energy')
41. plt.title('VQE PLOT')
42. plt.show()
该示例对应的输出结果如下,曲线图是氢分子在不同距离下对应的基态能量:
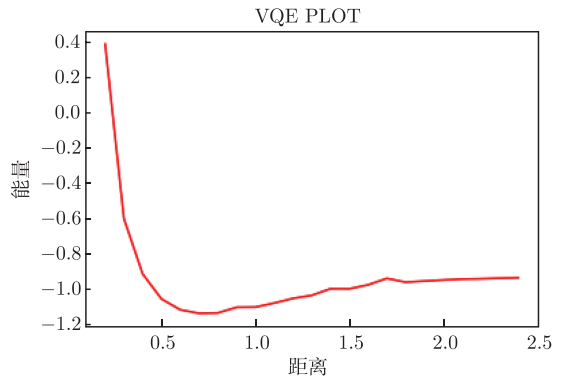
图附3.3.2 氢分子在不同距离下对应的基态能量